
Evaluate High-throughput Sequencing Reads with FastQC¶
Goal¶
The FastQC software is a popular way to evaluate the quality of high-throughput sequencing reads (e.g. reads from Illumina or PacBio sequencing). This quickstart won’t go into all of the nuances of interpreting these results (see instead the official FastQC Documentation ). Rather, we will get you using the tool right away in the Discovery Environment.
Prerequisites¶
Downloads, access, and services¶
In order to complete this tutorial you will need access to the following services/software
Prerequisite Preparation/Notes Link/Download CyVerse account You will need a CyVerse account to complete this exercise Register
Platform(s)¶
We will use the following CyVerse platform(s):
| Platform | Interface | Link | Platform Documentation | Learning Center Docs |
|---|---|---|---|---|
| Data Store | GUI/Command line | Data Store | Data Store Manual | Guide |
| Discovery Environment | Web/Point-and-click | Discovery Environment | DE Manual | Guide |
Input and example data¶
In order to complete this quickstart you will need to have the following inputs prepared
| Input File(s) | Format | Preparation/Notes | Example Data |
|---|---|---|---|
| Sequencing reads | FastQ | Any sequencing reads in FastQ format will work. They do not need to be pre-processed. They may also be compressed (e.g. fastq.gz) | SRR1028781.fastq |
Get started: Evaluate Reads with FastQC¶
Tip
If you have not already imported your own sequence read files to CyVerse, you can follow the instructions for uploading data, for example using Cyberduck, in our Data Store guide
Login to the Discovery Environment.
Click FastQC 0.11.5 (multi-file) to open the App, or click on Apps in the DE workspace and search for and run FastQC 0.11.5.
Under “Analysis Name” leave the defaults or make any desired notes.
Under “Select Input data” for ‘Input file, click Browse, then navigate to and select one or more FastQ files to analyze; Then click OK.
Note
To use our example data, navigate to Community Data > cyverse_training > quickstarts > fastqc and select the SRR1028781.fastq file.
Click Launch Analysis. You will receive a notification and may close the Apps window.
Click on Analyses from the DE workspace and monitor the status of your submitted job (You may have to click refresh to view updated status).
In the Analysis console, once your status appears as ‘Completed,’ click on the name of your analysis to navigate you to the results. Download the result files (in zip format) using the simple download, unzip the files and open the results in a web browser.
Summary¶
Analyzing a FastQC report, you can evaluate the quality of your sequencing results. The best way to interpret this report is to consult the official FastQC Documentation. You should keep in mind that simply because individual reports may generate a warning or fail, this does not mean your data are unusable. In most cases poor quality reads can be eliminated by subsequent cleaning steps without losing a large amount of sequence. Some reports such as ‘Sequence Duplication Levels’ might generate a warning when analyzing RNA-Seq data where you have many highly expressed transcripts. Here are a few tips:
Tip
Here are some of the most important reports to consider in downstream cleaning steps. Having a fail on these reports would require careful evaluation of whether or not the data can be sufficiently cleaned to be useful. These tips may not apply in every situation, you will have to interpret or seek advice on your own results.
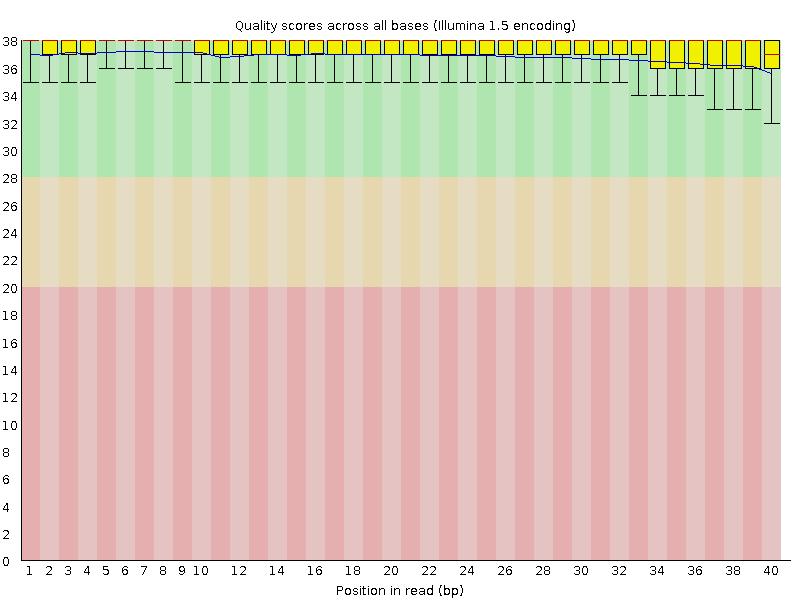
Per base sequence quality
This report shows the average quality score across the length of all reads. Poor quality at the beginning or end of the reads may suggest settings for trimming.
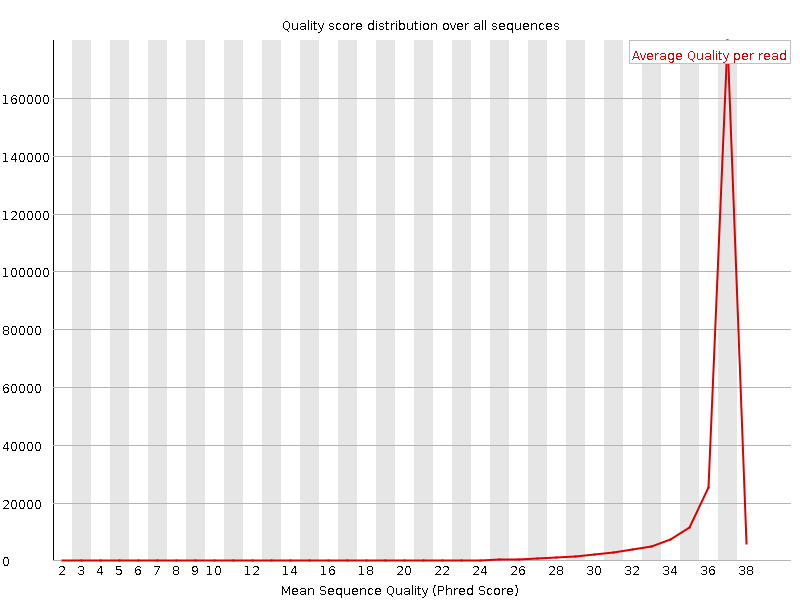
Per sequence quality scores
This report indicates how individual reads of a given quality score are distributed in your sequence file. Ideally, most reads will have a high average quality score. Populations of lower average-scored reads can be removed by downstream filtering.
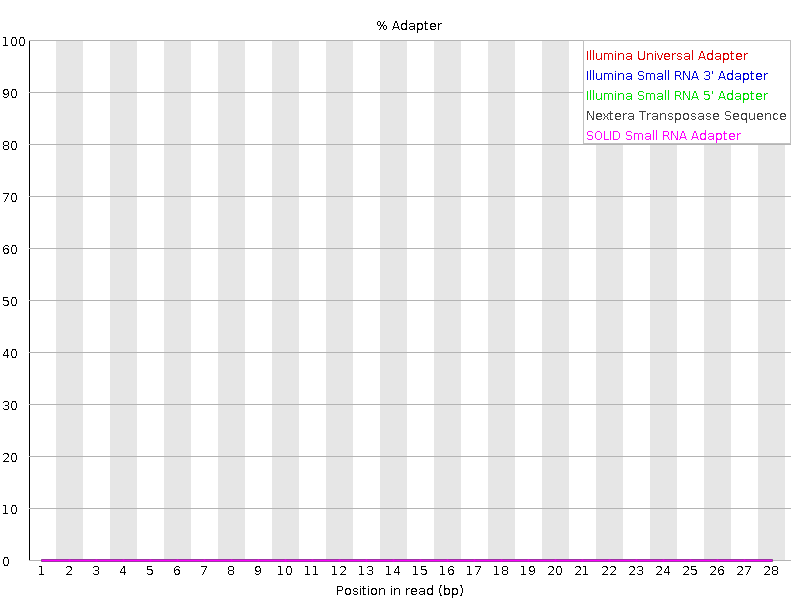
Adapter Content
This report indicates the presence of sequencing adapters. If adapters are detected, you will need to remove them in downstream cleaning.
Next Steps:
Following your report, you may wish to apply one of several tools in the Discovery Environment to, for example, remove sequencing adaptors and trim low quality portions of reads. The Trimmomatic-programmable-0.33 app is suggested.
Additional information, help¶
See the original FastQC Documentation for all the instructions on how to use this tool and interpret reports.
Fix or improve this documentation
- Search for an answer: |CyVerse Learning Center|
- Ask us for help: click |Intercom| on the lower right-hand side of the page
- Report an issue or submit a change: |Github Repo Link|
- Send feedback: Tutorials@CyVerse.org